Programmes et banques de données spectroscopiques


- kc_data:
- a:8:{i:0;s:0:"";s:4:"mode";s:2:"kc";s:3:"css";s:0:"";s:9:"max_width";s:0:"";s:7:"classes";s:0:"";s:9:"thumbnail";s:0:"";s:9:"collapsed";s:0:"";s:9:"optimized";s:0:"";}
- kc_raw_content:
- [kc_row use_container="yes" _id="766724"][kc_column width="12/12" video_mute="no" _id="936058"][kc_column_text _id="607906"]
Programmes et banques de données spectroscopiques
[/kc_column_text][kc_spacing height="20" _id="216610"][/kc_column][/kc_row][kc_row use_container="yes" _id="925114"][kc_column width="12/12" video_mute="no" _id="749820"][kc_spacing height="50px" _id="756149"][/kc_column][/kc_row][kc_row use_container="yes" _id="839814"][kc_column width="2%" _id="668404"][/kc_column][kc_column width="40%" _id="155296"][kc_single_image image_size="full" _id="270807" image_source="media_library" image="13808"][kc_spacing height="20" _id="467470"][kc_column_text _id="536384"]{slide=XTDS MOLECULAR SPECTROSCOPY SOFTWARE}
The Dijon spectroscopy group has developed powerful techniques based on group theory and tensorial formalism [1,2] in order to analyze and simulate absorption and Raman spectra of molecules with various symmetries. Software packages and databases implementing these tools have been created:
- XY4 [3] (Td tetrahedral symmetry, STDS package),
for 12CH4, 13CH4, 12CD4, 13CD4, 116SnH4, 28SiH4, 29SiH4, 30SiH4, 28SiF4, 70GeH4, 72GeH4, 73GeH4, 74GeH4, 76GeH4, 70GeD4, 72GeD4, 73GeD4, 74GeD4, 76GeD4, 70GeF4, CF4, P4, Ni(CO)4, 184OsO4, 186OsO4, 187OsO4, 188OsO4, 189OsO4, 190OsO4, 192OsO4 - XY6 [4] (Oh octahedral symmetry, HTDS package),
for 182WF6, 183WF6, 184WF6, 186WF6, 80SeF6, 32SF6, 33SF6, 34SF6, 235UF6, 238UF6, Mo(CO)6 - XY2Z2 [5] (C2v symmetry, C2vTDS package),
for SO2F2 - XY5Z [6] (C4v symmetry, C4vTDS package),
for SF5Cl - X2Y4 [7] (D2h symmetry and D2hTDS package),
for C2H4 - XY3Z [8] (C3v symmetry, C3vTDS package for closed-shell systems and C3vSTDS package for open-shell systems),
for CH3D and CH3O
These packages all consist in FORTRAN programs called by scripts.
XTDS [9] is a user-friendly java-based interface, which allows to interactively build and launch spectrum calculation or analysis jobs using any of the above-mentioned packages. This software runs on UNIX (UNIX, Linux or Mac OS X computers) and Windows systems. It allows the treatment of complex spectroscopic problems, including molecules with complex polyads, like methane for instance. The user can define the polyad scheme for the molecule under consideration. All interaction terms up to a given order of the development are automatically determined for both the Hamiltonian or transition moments (dipole moment or polarizability). Least-squares fits of experimental data can also be run interactively.
The full XTDS software (al packages, Java graphical interface), or the individual packages lister above (in this case with no graphical interface) can be download through the links in the table below:
The full XTDS software (al packages, Java graphical interface), or the individual packages lister above (in this case with no graphical interface) can be download through the links in the table below:
XTDS :
Read me file : Unix
Download links : Unix (Intel)References:
- [1] V. Boudon, J.-P. Champion, T. Gabard, M. Loete, F. Michelot, G. Pierre, M. Rotger, Ch. Wenger and M. Rey, J. Mol. Spectrosc., 228, 620-634 (2003).
- [2] “ Spherical Top Theory and Molecular Spectra ”, V. Boudon, J.-P. Champion, T. Gabard, M. Loëte, M. Rotger and Ch. Wenger, In Handbook of High-Resolution Spectroscopy, M. Quack and F. Merkt Editors, vol. 3, pp 1437–1460, Wiley, 2011.
- [3] Ch. Wenger and J.-P. Champion, J. Quant. Spectrosc. Radiat. Transfer, 59, 471-480 (1998).
- [4] Ch. Wenger, V. Boudon, J.-P. Champion and G. Pierre, J. Quant. Spectrosc. Radiat. Transfer, 66, 1-16 (2000).
- [5] Ch. Wenger, M. Rotger and V. Boudon, J. Quant. Spectrosc. Radiat. Transfer, 93, 429-446 (2005).
- [6] Ch. Wenger, M. Rotger and V. Boudon, J. Quant. Spectrosc. Radiat. Transfer, 74, 621-636 (2002).
- [7] Ch. Wenger, W. Raballand, M. Rotger and V. Boudon, J. Quant. Spectrosc. Radiat. Transfer, 95, 521-538 (2005).
- [8] A. El Hilali, Ch. Wenger, V. Boudon and M. Loëte, J. Quant. Spectrosc. Radiat. Transfer, 111, 1305–1315 (2010).
- [9] Ch. Wenger, V. Boudon, M. Rotger, M. Sanzharov and J.-P. Champion, J. Mol. Spectrosc., 251, 102–113 (2008).
{/slide}
[/kc_column_text][kc_spacing height="50px" _id="932321"][kc_single_image image_size="full" _id="690047" image_source="media_library" image="13814"][kc_spacing height="20" _id="246861"][kc_column_text _id="768680"]{slide=MIRS MOLECULAR SPECTROSCOPY SOFTWARE}
The MIRS spectroscopic software for the modeling of ro-vibrational spectra of polyatomic molecules is designed for the global treatment of complex band systems of molecules to take full account of symmetry properties. It includes efficient algorithms based on the irreducible tensor formalism. Predictions and simultaneous data fitting (positions and intensities) are implemented as well as advanced options related to group theory algebra. Illustrative examples on CH3D, CH4, and PH3, complete projects on CH3D and CH3Cl are included in the present version.
Getting and Installing:
The MirsDistr.exe file is a self-extractive archive to be extracted into a proper Temp directory. The complete installation uses a standard assistant launched by the setup.exe file in the Tempdisk1 directory. MIRS written in C++ includes all necessary executable files for Windows 95 and higher. The CPU and disk space requirements obviously depend on the complexity of the problem to treat. The package itself requires a minimum of 15 Mb of free disk space. The total amount of disk space occupied when all the projects included in the package have been build is close to 500 Mb. A typical execution times using a Pentium 4 processor at 2.4 GHz is about 10 minutes to build the CH3D project (all matrix element and prediction files) and less than 60 minutes to build each of the CH3Cl projects (two isotopomeres).
Two versions of the Mirs distribution:
You can download two different versions of the Mirs distribution. The full version contains tables for examples calculation. The minimal version does not contains any big table files and you will need calculate full set of tables on your computer. For all examples except CH3Cl you need build tables up to J=25 (Tables:BuiltDefault:J25). For CH3Cl you need build tables up to J=80 (Tables:BuiltDefault:J80). The average time of tables calculation on Pentium 4 processor at 2.4 GHz is one, two hours.
- Click to download full version of Mirs MirsDistr.exe (26 Mb)
- Click to download minimal version of Mirs MirsDistrMin.exe (17 Mb)
Getting started:
Standard features and options are directly accessible from the tutorial projects in the examples directory in which all necessary modelling files are included. It is recommended to create a new working directory for each new project. The Help menu can be accessed at any stage of the project building and subsequent calculations.
In the File menu select the Open ... command to open the CH3D.MRS file in the examplesch3d.j4.fit directory. A graphical picture of the project is displayed in purple. Select the Build command from the Calculus menu to generate all files connected with this project. At the end of the process the graphical picture appears in green indicating that all files are created consistently. The results are stored in ASCII files in the examplesCH4 working directory. Refer to the Help command for details on the content of each file. To explore the other tutorial projects (CH3D_LIGHT; PH3_LIGHT) repeat the same commands from the corresponding working directories. For the full project CH3D_FULL and CH3Cl_FULL (12_35and 12_37) it is necessary to first extend to higher J values the calculation of coupling coefficients. This can be easily achieved from the Tables:BuiltDefault:J80 command of the Tools menu.
Important:
Several improvements are under construction to correct minor failures of the user interface. They will be included in due course in subsequent releases. For any question contact Andrei Nikitin (avn@lts.iao.ru).
{/slide}
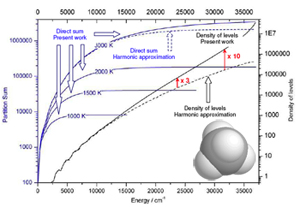
[/kc_column_text][kc_spacing height="50px" _id="299990"][kc_single_image image_size="full" _id="47402" image_source="media_library" image="13819"][kc_spacing height="20" _id="959105"][kc_column_text _id="141039"]{slide=THE METHANE PARTITION FUNCTION}
To calculate de methane partion function at a given temperature, click on this link.
{/slide}
[/kc_column_text][/kc_column][kc_column width="16%" _id="501538"][/kc_column][kc_column width="40%" _id="234057"][kc_single_image image_size="full" _id="216248" image_source="media_library" image="18318"][kc_spacing height="20" _id="310122"][kc_column_text _id="39692"]{slide=SPVIEW SOFTWARE TO ASSIGN & ANALYZE MOLECULAR SPECTRA}
Sample TextaSPVIEW is a program that facilitates the analysis of high-resolution molecular spectra by allowing the user to assign lines using a graphical environment. With SPVIEW, it is possible to:
- Load experimental and simulated spectra as XY ASCII files.
- Create peak lists from experimental spectra.
- Load predicted line lists in various formats (XY, HITRAN, TDS).
- Manipulate the plots (zoom, rescale, move, ...).
- Assign/unassign lines graphically (with the mouse).
- Create local simulations for a limited wavenumber range.
- ... and more !
SPVIEW is written in Java and Fortran 77.
Reference:
To download SPVIEW:
SPVIEW :
Download links : All platforms{/slide}
[/kc_column_text][kc_spacing height="40px" _id="440976"][kc_single_image image_size="full" _id="246889" image_source="media_library" image="13816"][kc_column_text _id="232465"]{slide=THE GROUP PROGRAM}
The GROUP package intends to provide access to the G orientation matrix elements for the symmetrization of O(3) (or SU(2) x CI) tensors into the Td (or TdS) subgroup. It also lists various coefficients necessary for tensor algebra in these groups. These cofficients are necessary for spectroscopic calculations for tetrahedral XY4 and octahedral XY6 molecules. They are used in the STDS and HTDS packages. It is designed for UNIX machines. It consists in FORTRAN (ANSI 77) routines.
Main GROUP characteristics :
GROUP handles J values up to 199.5
It works for both integer and half integer J values. Half integer J values are necessary for handling systems with an odd number of electrons.References:
- [1] J.-P. Champion, G. Pierre, F. Michelot and J. Moret-Bailly, Can. J. Phys., 55, 512-520 (1977).
- [2] V. Boudon and F. Michelot, J. Mol. Spectrosc., 165, 554-579 (1994).
- [3] M. Rey, V. Boudon, Ch. Wenger, G. Pierre and B. Sartakov, J. Mol. Spectrosc., 219, 313-325 (2003).
To download GROUP:
Read me files : Unix
Download links : Unix (Intel){/slide}
[/kc_column_text][/kc_column][kc_column width="2%" _id="154268"][/kc_column][/kc_row] - XY4 [3] (Td tetrahedral symmetry, STDS package),
- auto_update_uri:
- -1